
![]()
При посиланні на статтю з нашого журналу
необхідно вказувати:
- назву журналу
українською мовою - Медичні перспективи
або транслітерацією - Medicni perspektivi
- DOI статті
![]()


Медичні перспективи. 2026. Т. 31, № 1
2026 Том XXXI № 1
Опубліковано
31-03-2026
Ключові слова: громадське здоров’я, симуляційні технології, готовність до надзвичайних ситуацій, національна безпека, кризове реагування, симуляційне навчання
Key words: public health, simulation technologies, emergency preparedness, national security, crisis response, simulation-based training
Реферат
Система громадського здоров’я України функціонує в умовах поєднання воєнних загроз, інтенсивних міграційних процесів, трансформації соціального середовища та зростання ризиків біологічних, хімічних і техногенних інцидентів. За таких обставин традиційні освітні й управлінські підходи є обмеженими для формування готовності до комплексних кризових ситуацій і не дозволяють повною мірою оцінити спроможність системи до скоординованого реагування. У відповідь на ці виклики симуляційні технології дедалі частіше розглядаються як інструмент, що поєднує підготовку персоналу з тестуванням управлінських рішень і міжвідомчої взаємодії в безпечному середовищі. Метою роботи є концептуально-аналітичне узагальнення сучасних підходів до використання симуляційних технологій у сфері громадського здоров’я та формування прикладної рамки їх інтеграції для підвищення готовності й функціональної стійкості національної системи громадського здоров’я України до сучасних і комбінованих загроз. Аналіз ґрунтувався на опрацюванні рецензованих наукових публікацій, міжнародних методологічних рекомендацій і стратегічних документів, відібраних у міжнародних наукометричних базах даних та на офіційних ресурсах провідних міжнародних інституцій, переважно за період 2018-2025 років. Обґрунтовано доцільність розгляду симуляційних технологій не лише як освітнього інструмента, а як складової забезпечення системної готовності та функціональної стійкості громадського здоров’я. Систематизовано основні напрями застосування симуляцій, зокрема реагування на надзвичайні ситуації, тестування управлінських політик, кризову комунікацію та міжвідомчу координацію. Узагальнено роль медичних університетів як інституційної основи розвитку симуляційної підготовки та ідентифіковано ключові бар’єри її впровадження, включно з інфраструктурними, нормативними й методичними обмеженнями. На підставі отриманих аналітичних узагальнень сформульовано стратегічні напрями розвитку симуляційних технологій у системі громадського здоров’я України, реалізація яких розглядається як передумова підвищення готовності системи до сучасних і майбутніх загроз.
Abstract
Simulation as a tool for strengthening the preparedness of Ukraine’s public health system to threats: challenges and prospects. Miasoiedov V.V., Merkulova T.V., Nesterenko V.G., Peresypkina T.V. The public health system of Ukraine operates under a combination of military threats, intensive migration processes, transformation of the social environment, and increasing risks of biological, chemical, and technological incidents. Under these conditions, traditional educational and managerial approaches are limited in their capacity to ensure preparedness for complex crisis situations and do not allow for a comprehensive assessment of the system’s ability to respond in a coordinated manner. In response to these challenges, simulation technologies are increasingly considered as a tool that integrates workforce training with the testing of managerial decisions and intersectoral coordination in a safe environment. The aim of this study is a conceptual and analytical synthesis of contemporary approaches to the use of simulation technologies in public health and the development of an applied framework for their integration to enhance the preparedness and functional resilience of the national public health system of Ukraine to modern and combined threats. The analysis was based on peer-reviewed scientific publications, international methodological guidelines, and strategic documents retrieved from international scientific databases and official resources of leading international institutions, primarily covering the period from 2018 to 2025. The study substantiates the relevance of considering simulation technologies not only as an educational tool but as a component of ensuring systemic preparedness and functional resilience in public health. The main areas of application of simulations are systematized, including emergency response, testing of public policies, crisis communication, and intersectoral coordination. The role of medical universities as an institutional foundation for the development of simulation-based training is summarized, and key barriers to implementation are identified, including infrastructural, regulatory, and methodological constraints. Based on the analytical synthesis, strategic directions for the development of simulation technologies in the public health system of Ukraine are formulated, the implementation of which is considered a prerequisite for strengthening system preparedness for current and future threats.
Сучасні умови функціонування системи громадського здоров’я України формуються під впливом поєднання воєнних загроз, інтенсивних міграційних процесів, трансформації соціального середовища та зростання ризиків біологічних, хімічних і техногенних інцидентів [1]. За таких обставин традиційні підходи до підготовки фахівців й оцінювання інституційної готовності є обмеженими, оскільки не відтворюють багатофакторний і динамічний характер сучасних загроз. У відповідь на це в країнах з розвиненими системами безпеки дедалі ширше застосовуються симуляційні технології, які дозволяють моделювати кризові сценарії без ризику для населення та інфраструктури, поєднуючи підготовку персоналу з тестуванням управлінських механізмів [2, 3].
У сучасній практиці симуляція виходить за межі суто освітнього інструмента й розглядається як компонент забезпечення функціональної готовності систем громадського здоров’я та національної безпеки. Використання симуляцій дає змогу оцінювати реалістичність алгоритмів реагування, виявляти прогалини в міжвідомчій координації, перевіряти управлінські рішення та формувати поведінкові компетентності, необхідні для роботи в умовах невизначеності [4, 5, 6, 3]. Аналітичні огляди й дослідження у сфері громадського здоров’я та управління надзвичайними ситуаціями підтверджують доцільність розгляду симуляції як інструмента доказового управління, що дозволяє оцінювати ефективність організації реагування до настання реальних подій [7, 2, 3].
Важливим є також поведінковий і психоемоційний аспект симуляційного навчання. Дані міжнародних досліджень свідчать, що участь у симуляційних тренінгах підвищує впевненість фахівців, покращує командну взаємодію та знижує рівень тривожності в кризових ситуаціях, що безпосередньо впливає на якість управлінських рішень [8, 9]. Для України, де система громадського здоров’я одночасно реагує на наслідки воєнних дій, цивільні надзвичайні ситуації та загрози біобезпеці, ці характеристики набувають особливого значення.
У статті проаналізовано можливості та обмеження впровадження симуляційних технологій у систему громадського здоров’я України, обґрунтовано їх стратегічну роль, визначено потенціал університетів і міжнародного партнерства у формуванні національної моделі симуляційної підготовки, а також окреслено ризики, що потребують нормативного й наукового врегулювання.
Метою роботи є узагальнення сучасних підходів до використання симуляційних технологій у сфері громадського здоров’я як інструмента системної готовності та функціональної стійкості, а також формування прикладної концептуальної рамки інтеграції симуляцій у національну систему громадського здоров’я України в умовах сучасних і комбінованих загроз із визначенням ключових стратегічних напрямів їх подальшого розвитку.
МАТЕРІАЛИ ТА МЕТОДИ ДОСЛІДЖЕНЬ
Роботу виконано у форматі концептуально-аналітичного огляду проблеми з елементами картування доказів. Дизайн обрано з огляду на фрагментарність наявних наукових публікацій щодо використання симуляції саме в системі громадського здоров’я: більшість досліджень зосереджені або на клінічній симуляції, або на окремих компонентах готовності, тоді як комплексні емпіричні дослідження на стику «симуляція – громадське здоров’я – системна готовність» представлені обмежено. Методологічний підхід спрямований на узагальнення наявних наукових і нормативно-методичних джерел, їх тематичний аналіз та формування прикладної концептуальної рамки, релевантної для умов України.
Пошук літератури здійснено в міжнародних наукових базах даних PubMed, Scopus та Web of Science; додатково проведено цільовий пошук керівництв, практичних настанов і стратегічних документів на офіційних ресурсах міжнародних інституцій (зокрема ВООЗ і Європейського центру профілактики і контролю захворювань) та в національних нормативних і стратегічних документах України.
Для формування вибірки використовувались комбіновані пошукові запити українською та англійською мовами, анотації та ключові слова, які охоплювали теми громадського здоров’я, безпеки здоров’я, готовності та стійкості системи охорони здоров’я, симуляційних вправ, включно з післядієвим аналізом, а також компоненти підготовки кадрів, управлінських рішень, політик, координації та кризових комунікацій; окремо застосовано уточнювальні запити, пов’язані з підходом «Єдине здоров’я» (One Health). Часовим інтервалом відбору джерел були 2018-2025 рр., з можливістю включення окремих базових концептуальних джерел, опублікованих раніше. З вибірки виключено роботи, що описували виключно клінічну симуляцію без зв’язку із системною готовністю та управлінням у громадському здоров’ї, освітні або інформаційні матеріали без методичного обґрунтування й аналітичної цінності, дублікати, а також короткі повідомлення або новини без емпіричних чи аналітичних даних. Узагальнення виконували шляхом тематичного синтезу й рамкового аналізу з опорою на цикл готовності «підготовка – реагування – оцінка – покращення». Для кожного джерела структуровано вилучали інформацію про тип симуляційної вправи, мету застосування, рівень системи, наявність формалізованого оцінювання та потенціал адаптації до умов України.
Систематизацію джерел й описове картування (розподіл за типом джерела, тематичними напрямами та роком публікації) виконано із застосуванням електронних таблиць (Microsoft Excel/Google Sheets); статистичні гіпотези не тестувалися, оскільки дизайн роботи є концептуально-аналітичним оглядом.
Дослідження не передбачало залучення людей як об’єктів дослідження, використання персональних або чутливих медичних даних, у зв’язку з цим етичне схвалення не вимагалося.
РЕЗУЛЬТАТИ ТА ЇХ ОБГОВОРЕННЯ
Результати дослідження ґрунтуються на тематичному синтезі та рамковому аналізі відібраних джерел і відображають концептуальні підходи та закономірності застосування симуляцій у системі громадського здоров’я, а не кількісну оцінку ефектів чи показників.
У межах цієї роботи ключові поняття використовуються в такому значенні. Під готовністю системи громадського здоров’я розуміється спроможність інституцій, кадрового потенціалу та організаційних процесів завчасно планувати, здійснювати підготовку і тренування, реалізовувати реагування та забезпечувати відновлення функцій під час або після надзвичайних подій і загроз із застосуванням циклу безперервного покращення, зокрема через симуляційні вправи та післядієвий аналіз із подальшими планами удосконалення [2].
Функціональна стійкість системи громадського здоров’я в цій статті розглядається як здатність системи зберігати критично важливі функції – епідеміологічний нагляд, лабораторну діагностику, координацію, комунікацію та ухвалення управлінських рішень – в умовах підвищеного навантаження, а також оперативно відновлювати їх, використовуючи уроки, отримані під час симуляційних вправ або реальних подій.
Симуляція або симуляційна вправа в громадському здоров’ї визначається як структурована діяльність з моделювання сценаріїв загроз, що реалізується у форматі настільних, функціональних або повномасштабних вправ і призначена для підготовки та/або перевірки компонентів готовності системи з обов’язковим етапом оцінювання результатів і подальшого покращення.
Термін «симуляційна модель» у межах цього дослідження використовується як концептуальна рамка проєктування та вбудовування симуляційних вправ у цикл готовності системи громадського здоров’я.
Інтеграція симуляцій у громадське здоров’я: від освітньої функції до інструмента державної готовності
Інтеграція симуляцій у практику громадського здоров’я розширює можливості підготовки персоналу порівняно з традиційними освітніми форматами, оскільки забезпечує відпрацювання поведінкових й управлінських навичок, критично важливих для готовності системи до реагування [2, 8]. На відміну від лекційних і семінарських занять, симуляційні вправи відтворюють ситуації дефіциту часу та невизначеності, у яких фахівці мають приймати рішення, координувати командну взаємодію, застосовувати алгоритми кризових комунікацій і діяти в умовах динамічної зміни обстановки [3, 5, 6]. Це робить симуляцію ефективним інструментом підготовки кадрів громадського здоров’я та суміжних секторів, зокрема епідеміологів, фахівців первинної ланки, управлінців, лабораторних працівників, представників місцевого самоврядування та комунікаційних команд [10, 3].
Український контекст зумовлює потребу в сценаріях, що виходять за межі класичних інфекційних спалахів і враховують комбіновані загрози, у яких поєднуються хімічні, радіаційні, техногенні та біологічні компоненти. В умовах військової агресії, яка триває, симуляції дозволяють моделювати ситуації, у яких рішення ухвалюються за високого рівня невизначеності та стресу, а також перевіряти функціональну стійкість системи громадського здоров’я за одночасної дії кількох типів небезпек. До таких сценаріїв належать, зокрема, інфекційний спалах в умовах масової евакуації або руйнування критичної інфраструктури, а також комбіновані події, що потребують скоординованих дій різних служб і секторів [11, 12, 13]. У цьому сенсі симуляційні вправи виконують не лише освітню, а й інструментальну функцію, як механізм перевірки операційної готовності та міжвідомчої взаємодії в умовах кризових навантажень [14, 2].
Узагальнення ролі симуляцій у системі громадського здоров’я подано у вигляді концептуальної моделі (рис.), що поєднує лінійний та циклічний виміри. Лінійний компонент відображає логіку переходу від контексту загроз до результатів на рівні готовності та функціональної стійкості системи, тоді як циклічний – механізм безперервного покращення через симуляційні вправи, оцінювання та корекцію управлінських рішень.
Рисунок. Концептуальна модель ролі симуляцій у формуванні готовності та функціональної стійкості системи громадського здоров’я ↓
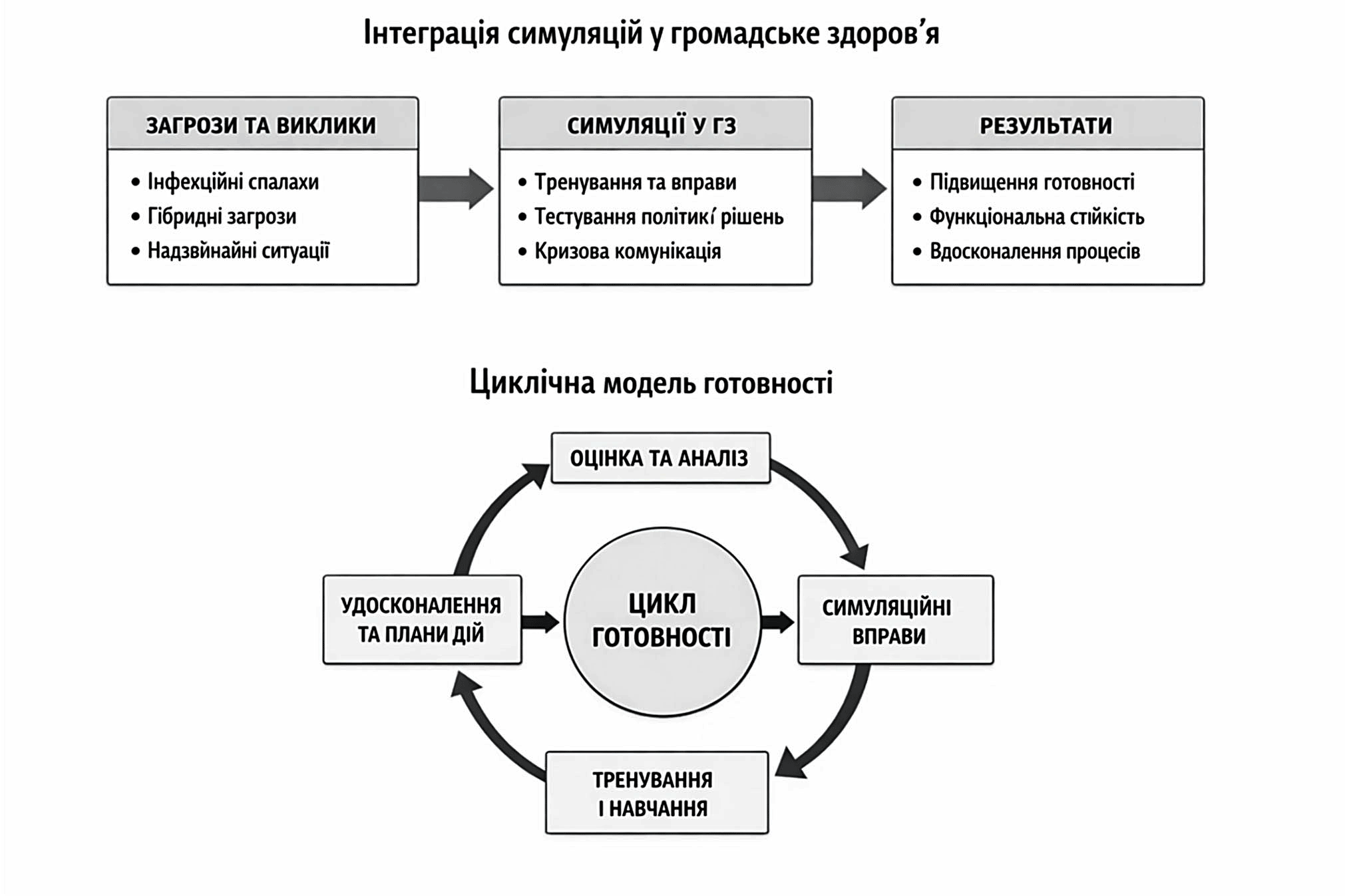
У міжнародній практиці симуляція також використовується як метод оцінювання операційної сумісності між службами. Методологічні документи Всесвітньої організації охорони здоров’я та Європейського центру профілактики і контролю захворювань передбачають застосування настільних, функціональних і повномасштабних вправ для перевірки ефективності системи реагування [3, 4, 5]. В Україні ці підходи поки застосовуються обмежено, переважно в контексті проєктних активностей або окремих навчань. Водночас регулярність симуляційних вправ є умовою підтримання узгодженості дій між структурами, які в повсякденній практиці взаємодіють нерегулярно, та підвищення готовності системи до скоординованого реагування [2, 3, 14].
Симуляція як інструмент формування поведінкових й управлінських компетентностей
Фахові дослідження та огляди останніх років у галузі симуляційної педагогіки, кризового менеджменту та підготовки фахівців громадського здоров’я свідчать, що симуляційні тренінги впливають насамперед на поведінкові та управлінські компетентності, критично важливі для ефективного кризового менеджменту, зокрема стратегічне мислення, лідерство, ухвалення рішень у стресових умовах, кризову комунікацію, орієнтацію на результат і толерантність до невизначеності [15, 16, 7, 8, 9]. Формування цих компетентностей відбувається переважно в середовищі, наближеному до реальних умов реагування, що обмежено або недосяжно в межах традиційних освітніх форматів.
Особливо значущою є роль симуляцій у підготовці епідеміологічних і лабораторних фахівців, діяльність яких пов’язана з раннім виявленням загроз, проведенням епідемічних розслідувань, визначенням випадку, управлінням даними та кризовими комунікаціями [10, 15]. Симуляційні вправи дозволяють відпрацювати повну траєкторію реагування – від фіксації первинних сигналів до ухвалення управлінських рішень щодо запровадження обмежувальних заходів або інформування населення – у логіці, наближеній до реального функціонування системи [3-5].
Вагомим компонентом впливу симуляцій є також формування психологічної готовності фахівців. Дані досліджень показують, що тренінги з елементами стрес-моделювання сприяють підвищенню впевненості, зменшенню тривожності та покращенню здатності працювати в багатопрофільних командах [8, 9]. В умовах тривалих кризових навантажень і війни цей аспект набуває стратегічного значення, оскільки безпосередньо впливає на здатність системи громадського здоров’я зберігати працездатність і підтримувати узгоджені дії персоналу.
Медичні університети як ядро розвитку симуляційних технологій
Система медичної освіти України розглядається як одна з ключових інституційних основ для розвитку національної моделі симуляційної підготовки у сфері громадського здоров’я. На відміну від відомчих структур, медичні університети поєднують освітню, наукову та методичну функції, що створює умови для системного впровадження симуляційних технологій. Університети можуть забезпечувати підготовку тренерів за моделлю підготовки інструкторів (train-the-trainer), розроблення та апробацію симуляційних сценаріїв, науковий супровід безпечності й ефективності симуляційного середовища, а також формування кадрового резерву для системи безперервного професійного розвитку фахівців [17, 18, 5].
Заклади вищої освіти мають можливість інтегрувати симуляційні технології в освітні компоненти програм медичних і медико-соціальних спеціальностей, забезпечуючи формування компетентностей – від базових клінічних і комунікаційних до навичок міжвідомчої взаємодії у складних кризових сценаріях [8, 16, 18]. Такий підхід узгоджується з європейськими моделями підготовки, у яких симуляція розглядається як невід’ємний елемент освіти та підвищення кваліфікації фахівців, залучених до охорони здоров’я та управління ризиками [19, 3].
Міжнародний досвід: можливості адаптації для України
У міжнародній практиці симуляційні методи розглядаються як інструмент підвищення готовності та запобігання кризам, а не лише як елемент навчання. У низці країн симуляційні центри та програми використовуються для тестування управлінських рішень, міжпрофесійної взаємодії та операційної сумісності служб у кризових сценаріях [2, 3]. У Європейському Союзі симуляційні вправи інтегровані в механізми оцінювання готовності систем громадського здоров’я, а міждержавні симуляційні вправи (Simulation Exercises, SimEx), що регулярно проводяться Європейським центром з профілактики та контролю захворювань, слугують інструментом перевірки координації та узгодженості реагування [20, 21]. На глобальному рівні підходи ВООЗ передбачають використання результатів симуляцій і післядієвого аналізу для корекції політик і планів реагування на основі принципу ідентифікованих уроків (lessons identified) [4]. У сукупності ці практики формують рамку, що може бути адаптована в Україні, зокрема в частині створення міжвідомчих платформ для спільного навчання, аналізу рішень і підвищення операційної готовності.
Окремий аналітичний інтерес для України становить модель One Health, яка ґрунтується на міжсекторальній взаємодії систем охорони здоров’я, ветеринарної медицини та охорони довкілля і підтримується провідними міжнародними організаціями. Практика проведення One Health Simulation Exercises демонструє ефективність спільного відпрацювання сценаріїв для служб громадського здоров’я, безпеки харчових продуктів і ветеринарії, особливо в умовах зоонозних і комбінованих загроз [22, 11, 12, 13]. Наявний в Україні базовий досвід міжсекторальних тренінгів за підтримки ВООЗ і партнерських структур створює передумови для подальшого інституційного закріплення та масштабування таких підходів на національному рівні [4, 5].
Бар’єри впровадження та ризики, що потребують системної відповіді
Інтеграція симуляційних технологій у систему громадського здоров’я України стримується не окремими ізольованими проблемами, а сукупністю структурних і нормативних обмежень, які формують системний бар’єр для їх масштабування та сталого використання. Ці обмеження стосуються інфраструктурного забезпечення, регуляторного середовища та механізмів оцінювання результатів симуляційної підготовки.
Однією з ключових проблем є нерівномірний розвиток симуляційної інфраструктури та обмежений доступ до відповідних ресурсів у регіонах, що ускладнює формування єдиного підходу до підготовки кадрів і міжвідомчої взаємодії. Фрагментарність цифрових рішень і відсутність інтегрованих платформ додатково знижують можливості координації, накопичення результатів та аналітичного використання даних симуляційних вправ [23, 14].
Вагомим регуляторним викликом залишається відсутність нормативно визначених вимог до безпечності та якості симуляційного середовища. Зокрема, в Україні не сформовано чітких стандартів щодо гігієнічних, психофізіологічних і когнітивних навантажень, пов’язаних з моделюванням критичних ситуацій. Із розширенням використання технологій віртуальної та доповненої реальності ці прогалини набувають додаткового значення, оскільки зростають ризики емоційної небезпеки, когнітивного перевантаження та ергономічних порушень, які наразі недостатньо досліджені й регламентовані [24, 25, 26].
Окремим системним обмеженням є відсутність уніфікованого підходу до оцінювання результатів симуляційних тренінгів і державного обліку їх проходження. Це унеможливлює повноцінну інтеграцію симуляцій у національну систему безперервного професійного розвитку, а також обмежує використання результатів симуляцій для планування кадрової політики, оцінювання готовності та обґрунтування управлінських рішень у сфері громадського здоров’я [4, 5].
Цифрова інфраструктура симуляційної підготовки
Формування національної моделі симуляцій у громадському здоров’ї потребує розвитку цифрової інфраструктури, яка забезпечує не лише проведення симуляційних вправ, а й їх координацію, документування та аналітичне використання результатів. У цьому контексті цифрові рішення розглядаються як інструмент масштабування симуляцій і підтримки системної готовності, а не як окремий технологічний компонент.
Ключовим елементом такої інфраструктури може бути віртуальна симуляційна платформа з національним репозиторієм сценаріїв, що дозволяє організовувати віддалені та змішані формати симуляцій, уніфікувати сценарії, накопичувати результати вправ і забезпечувати порівнюваність підходів до підготовки фахівців [4, 25, 26]. Наявність спільного цифрового середовища створює умови для узгодженого використання симуляційних сценаріїв і результатів між освітніми, науковими та практичними інституціями, залученими до функціонування системи громадського здоров’я [3, 18].
Окрім освітньої складової, цифрова інфраструктура розширює можливості використання симуляцій як інструмента аналізу та тестування управлінських рішень. Моделювання сценаріїв у цифровому середовищі дозволяє оцінювати потенційні наслідки управлінських алгоритмів і політичних рішень до їх реалізації в реальних умовах, що відповідає міжнародним практикам доказового ухвалення рішень у сфері громадського здоров’я та кризового управління [2, 19].
Стратегічні напрями розвитку симуляційних технологій у громадському здоров’ї
Подальший розвиток симуляційних технологій у сфері громадського здоров’я доцільно розглядати як перехід від фрагментарних освітніх ініціатив до цілісної, стандартизованої та науково обґрунтованої національної моделі, інтегрованої в систему підготовки кадрів, оцінювання готовності та формування політик. Узагальнення виявлених бар’єрів і міжнародних підходів дозволяє окреслити ключові стратегічні напрями такого розвитку.
Першим стратегічним напрямом є формування модульної системи симуляційної підготовки для різних категорій фахівців громадського здоров’я та суміжних секторів. Модульність дозволяє поєднувати базові та поглиблені сценарії реагування на інфекційні, техногенні, хімічні, радіаційні й комбіновані загрози з урахуванням професійних ролей і рівнів відповідальності.
Другим напрямом є інтеграція симуляцій у систему безперервного професійного розвитку як інструмента не лише навчання, а й перевірки та підтримання професійної готовності. Використання результатів симуляційних вправ у межах офіційних програм підвищення кваліфікації створює передумови для формування прогнозованої моделі розвитку кадрового потенціалу.
Третім стратегічним напрямом є розвиток національної мережі тренерів за моделлю «train-the-trainer». Підготовка сертифікованих інструкторів, здатних забезпечувати проведення симуляцій, адаптацію сценаріїв і методичний супровід у регіонах, є необхідною умовою масштабування симуляційних технологій і забезпечення рівного доступу до них.
Четвертий напрям стосується формування методичної та нормативної бази симуляційної підготовки. Йдеться про розроблення уніфікованих вимог до структури симуляційних вправ, критеріїв оцінювання результатів і стандартів безпечності симуляційного середовища, що забезпечують порівнюваність підходів і підвищення якості підготовки.
П’ятим напрямом є науковий супровід розвитку симуляційних технологій. Систематичні дослідження ефективності різних форматів симуляцій, валідності інструментів оцінювання та впливу симуляційного середовища на психофізіологічний стан учасників формують доказову основу для вдосконалення практик і стандартів.
Шостий стратегічний напрям пов’язаний з розвитком цифрових інструментів симуляційної підготовки, включно з віддаленими, змішаними та віртуальними форматами. Цифрові рішення розглядаються як інфраструктурна основа для масштабування симуляцій, уніфікації сценаріїв й аналітичного використання результатів.
Сьомим напрямом є посилення міжвідомчої координації шляхом впровадження симуляційних вправ за участю не лише системи охорони здоров’я, а й служб надзвичайних ситуацій, лабораторних, освітніх і соціальних інституцій. Такий підхід сприяє формуванню узгоджених алгоритмів реагування та підвищує здатність системи громадського здоров’я діяти в умовах комплексних криз.
Запропоновані стратегічні напрями узгоджуються із сучасними міжнародними підходами до використання симуляцій як інструмента підвищення готовності та функціональної стійкості систем громадського здоров’я й можуть слугувати основою для подальшого розвитку національної моделі симуляційної підготовки.
Обмеження дослідження: дослідження має обмеження, пов’язані з його концептуально-аналітичним характером, оскільки висновки ґрунтуються на узагальненні наукових і методичних джерел та не передбачають емпіричної перевірки або кількісної оцінки ефективності симуляційних інтервенцій.
ВИСНОВКИ
Проведений аналіз дозволив сформулювати такі узагальнення:
Внески авторів:
М’ясоєдов В.В. – концептуалізація, адміністрування проєкту;
Меркулова Т.В. – методологія, дослідження, написання – початковий проєкт;
Нестеренко В.Г. – формальний аналіз, курація даних;
Пересипкіна Т.В. – написання – рецензування та редагування, візуалізація.
Фінансування. Дослідження не має зовнішніх джерел фінансування.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів.
REFERENCES
Key words: hard capsules, quality control, Ishikawa diagram, critical parameters, technology
Ключові слова: тверді капсули, контроль якості, діаграма Ішікави, критичні параметри, технологія
Abstract
This article explores the application of the Ishikawa (cause-and-effect) diagram as a risk analysis tool within a pharmaceutical quality system for the production of hard capsules under the code name "Praziquantel Plus". Aligned with the standards of ISO 9001, ICH Q10, and GMP, the study demonstrates how this method systematically identifies and analyzes potential risks across key production categories. The research provides a tailored Ishikawa diagram that facilitates the identification of critical parameters, supports process optimization, and enhances the overall quality assurance system for this specific medicinal product. The aim of the research was to analyze the risks in the production of hard capsules under the conditional name “Praziquantel Plus” using the Ishikawa diagram. The study was based on the analysis of a model industrial process for the production of Praziquantel Plus hard capsules. The materials for the risk analysis were the technological regulations, specifications for raw materials and finished product, as well as the requirements of GMP/ICH standards. The study employed a systematic approach to risk assessment in accordance with ICH Q9 guidelines. The primary methodology is centered on the construction of the Ishikawa diagram. The study is based on a systems approach, brainstorming, cluster analysis and cause-and-effect analysis. A comprehensive Ishikawa diagram was developed, structured around six primary risk categories for pharmaceutical production: raw materials, production process, equipment, personnel, premises, and quality control. Critical risk factors were identified and prioritized. The most significant risks were found to be associated with: a) the quality of active pharmaceutical ingredients and excipients (e.g., inconsistency in quantitative content, impurities); b) critical stages of the technological process (e.g., weighing accuracy, mixture homogeneity, capsule filling flowability); and c) the specifics of pharmaceutical equipment qualification and operation. Detailed risk assessment tables were created for both raw materials and the technological process, outlining potential failures, their consequences, and proposed precautionary measures to mitigate each risk (e.g., enhanced incoming quality control, process validation, equipment calibration). An adapted, product-specific Ishikawa diagram was proposed for the industrial-scale production of “Praziquantel Plus”. This diagram details first- and second-order subcategories within the main "bones," reflecting the unique aspects of scaling up from laboratory to commercial manufacturing. The Ishikawa diagram method proves to be an effective and structured tool for pre-emptive risk identification in pharmaceutical process development and scale-up, aligning with GMP and ICH Q9/Q10 requirements. The study successfully mapped and systematized the key risk categories and their interrelations for the production of “Praziquantel Plus” hard capsules, providing a visual and analytical framework for quality management. The proposed tailored diagram and risk tables serve as a practical foundation for implementing preventive actions, optimizing the technological process, and conducting root-cause analysis of potential defects, thereby strengthening the overall Pharmaceutical Quality System for this product.
Реферат
Аналіз ризиків у виробництві твердих капсул під умовною назвою «Празиквантел плюс» методом діаграми Ішікави. Семченко К.В., Яковенко В.К., Шмалько О.О., Олійник С.В. У цій статті досліджується застосування діаграми Ішікави як інструменту аналізу ризиків у фармацевтичній системі якості для виробництва твердих капсул під умовною назвою «Празиквантел Плюс». Відповідно до стандартів ISO 9001, ICH Q10 та GMP, показано, як цей метод систематично виявляє та аналізує потенційні ризики в ключових категоріях виробництва. Запропоновано адаптовану діаграму Ішікави, яка полегшує ідентифікацію критичних параметрів, підтримує оптимізацію процесів та покращує загальну систему забезпечення якості для цього конкретного лікарського засобу. Мета цього дослідження полягала в аналізі ризиків у виробництві твердих капсул під умовною назвою «Празиквантел Плюс» за допомогою діаграми Ішікави. Дослідження ґрунтувалося на аналізі модельного промислового процесу виробництва твердих капсул «Празиквантел Плюс». Матеріалами для ризик-аналізу слугували технологічний регламент, специфікації на сировину та готовий продукт, а також вимоги стандартів GMP/ICH. У дослідженні було використано систематичний підхід до оцінювання ризиків відповідно до рекомендацій ICH Q9. Основна методологія зосереджена на побудові діаграми Ішікави. Дослідження базується на системному підході, мозковому штурмі, кластерному аналізі та причинно-наслідковому аналізі. Розроблено комплексну діаграму Ішікави, структуровану навколо шести основних категорій ризику для фармацевтичного виробництва: сировина, виробничий процес, обладнання, персонал, приміщення та контроль якості. Ідентифіковано та пріоритизовано критичні фактори ризику. Найбільш значущими виявилися ризики, пов'язані з: а) якістю АФІ та допоміжних речовин (напр., невідповідність кількісного вмісту, домішки); б) критичними етапами технологічного процесу (напр., точність зважування, однорідність суміші, сипкість маси при заповненні капсул); в) специфікою кваліфікації та експлуатації фармацевтичного обладнання. Створено детальні таблиці оцінки ризиків як для сировини, так і для технологічного процесу, де описані потенційні відмови, їх наслідки та запропоновані превентивні заходи для зменшення кожного ризику (напр., посилений вхідний контроль якості, валідація процесу, калібрування обладнання). Запропоновано адаптовану, продукт-специфічну діаграму Ішікави для промислового виробництва «Празиквантел Плюс». Ця діаграма деталізує підкатегорії першого та другого рівня в межах основних «кісток», що відображає особливості масштабування технології з лабораторного рівня на виробничий. Метод діаграми Ішікави зарекомендував себе як ефективний та структурований інструмент для превентивної ідентифікації ризиків при фармацевтичній розробці та масштабуванні процесів, що відповідає вимогам GMP та ICH Q9/Q10. Дослідження успішно систематизувало ключові категорії ризиків та їх взаємозв'язки для виробництва твердих капсул «Празиквантел Плюс», надавши візуальну та аналітичну основу для управління якістю. Запропоновані спеціалізована діаграма та таблиці ризиків слугують практичною основою для впровадження профілактичних дій, оптимізації технологічного процесу та аналізу основних причин потенційних дефектів, тим самим посилюючи загальну Фармацевтичну систему якості для цього препарату.
A Pharmaceutical Quality System (PQS) is a set of processes and procedures aimed at ensuring that pharmaceutical products are produced, stored and distributed in accordance with established quality standards. The basis of PQS is the international quality management system ISO 9001 and the requirements set out in the ICH Q10 guidelines. The system provides:
Quality of pharmaceutical products in Ukraine are regulated mainly by the requirements of State Pharmacopoeia of Ukraine, guidelines and the numerous orders of the Ministry of Health of Ukraine (MOHU) [1, 2, 3].
Risk analysis is a key component of PQS, which helps to minimize potential threats to product quality and patients. It is based on the principles outlined in the ICH Q9 guideline (“Quality Risk Management”). The main steps of risk analysis are risk identification, risk assessment, risk mitigation, risk monitoring and risk control.
Various methods are used to manage risks:
The Ishikawa diagram is an effective analytical tool for ensuring the quality of medicinal products. The main purpose of this method is to systematically identify and classify probable problems or causes of their occurrence in the production process. The use of the method is due to the complexity of the production of pharmaceutical products, when a combination of factors, such as the quality of incoming raw materials, equipment, technological methods, personnel, premises, etc., that can affect the quality of products both directly and indirectly.
The Ishikawa diagram gives the opportunity to structure the analysis process by highlighting the main categories of factors that can affect the quality of the medicinal product. This provides a comprehensive approach to solving problems, eliminating chaos in the search for causes. By detailing each category of factors, the root causes that lead to product defects, for example, uneven dosing of APIs in dosed preparations, can be identified. Usage of the diagram allows us to predict possible risks and create preventive measures aimed at improving production processes. This method admits enterprises to comply with high quality standards, such as the international Good Manufacturing Practice (GMP) standards and the international quality management system (ISO), ensuring quality control at all stages of production.
In scientific activities, the Ishikawa diagram construction method gives the opportunity to assess possible risks when scaling a laboratory technological process to a production one, establish possible critical parameters of the production process, and identify risks when developing quality control methods for medicinal products [4-8].
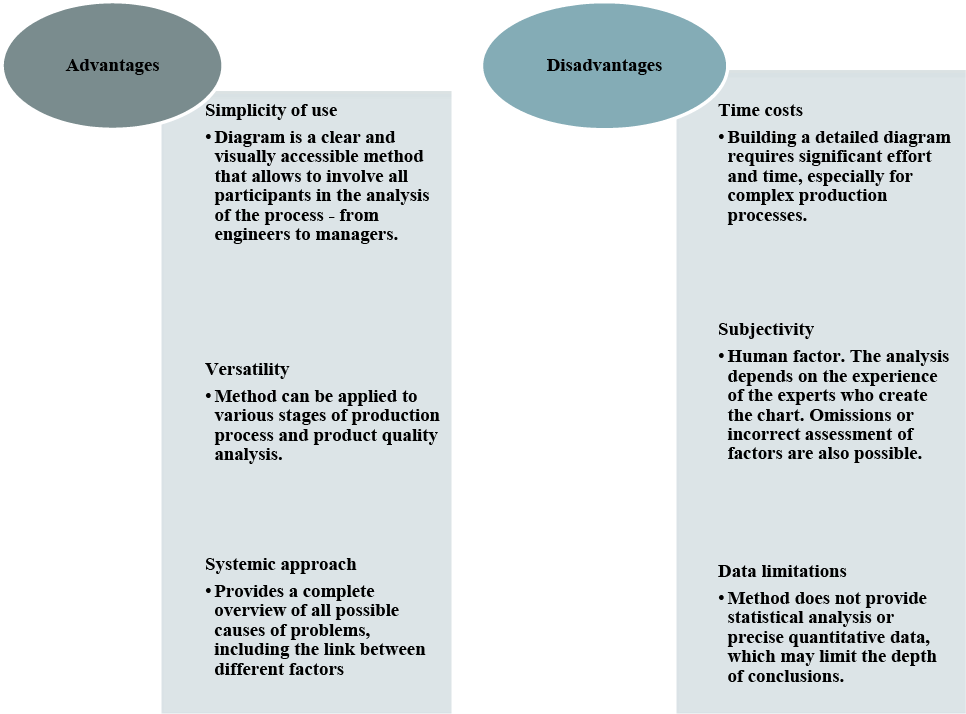
The Ishikawa diagram is an indispensable tool in pharmaceutical production, its usage contributes to the improvement of production processes, raising quality standards and preventing risks [9, 10]. However, it is important to consider the disadvantages of this method when combining it with other analytical tools for a deeper and more accurate analysis. The advantages and disadvantages of the Ishikawa diagram method are shown in Figure 1.
Fig. 1. Advantages and disadvantages of the Ishikawa diagram method ↓
The purpose of this study was to analyze the risks in the production of hard capsules under the conditional name “Praziquantel Plus” using the Ishikawa diagram.
MATERIALS AND METHODS OF RESEARCH
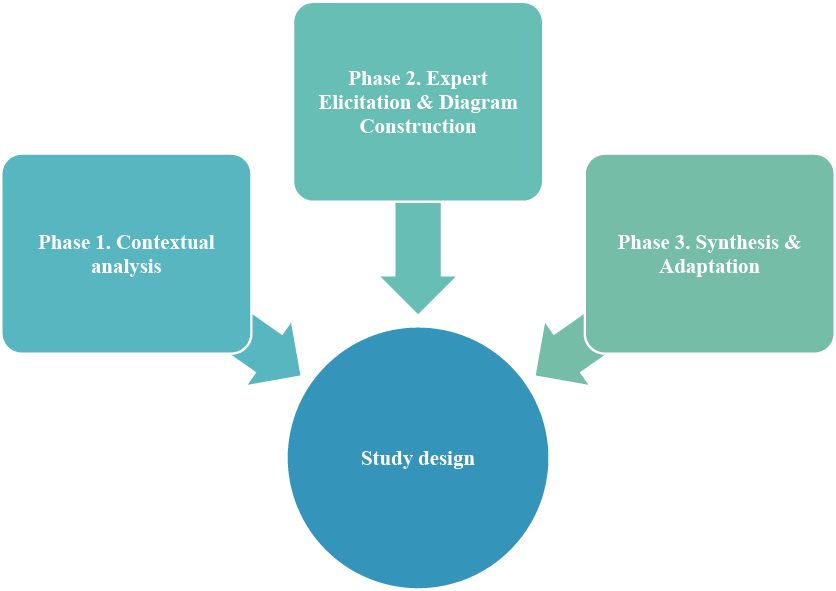
This research follows a qualitative, descriptive study design structured into three consecutive phases to ensure a systematic approach to risk identification and analysis (Fig. 2).
Phase 1: Contextual Analysis. A comprehensive review of the technological documentation for “Praziquantel Plus” capsules and relevant quality standards (GMP, ICH Q9, Q10) was conducted to define the scope and framework for risk assessment.
Phase 2: Expert Elicitation & Diagram Construction. The core analytical phase involved a structured brainstorming session with an expert panel (n=5) comprising pharmaceutical technologists, quality assurance, and production specialists. The elicited potential failure causes were then organized using cause-and-effect analysis and cluster analysis to construct a general Ishikawa diagram.
Phase 3: Synthesis & Adaptation. Based on the general diagram and a detailed process breakdown, a product-specific Ishikawa diagram for industrial-scale production was developed. Identified risks were further detailed and mitigation measures were proposed in tabular form.
Primary Analytical Tool. The Ishikawa (fishbone) diagram served as the primary visual and analytical tool throughout Phases 2 and 3 to structure and present the cause-and-effect relationships.
Fig. 2. Study design ↓
The basis for the study was the theoretical and information array necessary for modeling the risk analysis process. The materials of the study were a model technological process for the industrial production of hard capsules with combined content (code name “Praziquantel Plus”); a list of potential critical control points and quality parameters, identified based on the requirements of the State Pharmacopoeia of Ukraine, GMP and similar technologies; and categories of risk factors generally recognized in the pharmaceutical industry.
The study employed a systematic approach to risk assessment in accordance with ICH Q9 guidelines [2]. The primary qualitative methodology centered on the construction of an Ishikawa diagram. The process involved the following steps:
For risk prioritization, experts assessed each identified cause based on its estimated probability of occurrence and severity of impact on critical quality attributes. This qualitative assessment allowed for the identification of the most critical parameters requiring control measures, as presented in Tables 1 and 2.
For risk prioritization, a semi-quantitative approach based on the Risk Priority Number (RPN) was employed, in accordance with the principles of ICH Q9. Following the brainstorming session and construction of the general Ishikawa diagram, the expert panel (n=5) assessed each identified potential cause of failure.
Each risk was evaluated using two parameters:
Probability (P): The likelihood of the risk occurring.
Severity (S): The seriousness of the potential impact on the critical quality attributes (CQAs) of “Praziquantel Plus” capsules and patient safety.
Both parameters were scored on a 3-point ordinal scale (1=Low, 2=Medium, 3=High). The consensus scores were then multiplied to calculate the Risk Priority Number (RPN=P×S), which could range from 1 to 9.
Based on the RPN value, risks were prioritized for further analysis and mitigation:
High-priority (RPN≥6): Risks requiring immediate preventive actions and detailed control strategies (e.g., in-process controls, validation).
Medium-priority (RPN 3-4): Risks requiring monitoring and standard quality control procedures.
Low-priority (RPN 1-2): Acceptable risks under current control measures.
This prioritization enabled the research team to focus the subsequent detailed analysis on the “Raw Materials” and “Technological Process” categories, which contained over 70% of all high-priority risks.
Given the qualitative nature of the study and the use of expert elicitation data, formal statistical software was not employed for hypothesis testing. The semi-quantitative RPN calculation was performed using Microsoft Excel 365. Data organization and diagram structuring were supported by standard features of the software.
This study involved a structured expert elicitation session (brainstorming) as part of the risk analysis methodology. Participation was voluntary, and all experts provided informed verbal consent after being briefed on the study’s purpose and the anonymous, aggregated use of their input for academic publication. No personal or sensitive data were collected. The research procedure, focusing on process analysis rather than human subjects, was conducted in accordance with the ethical principles for non-interventional studies. Formal review by an institutional ethics committee was not required for this type of methodological research, as confirmed by the internal regulations of the National University of Pharmacy.
RESULTS AND DISCUSSION
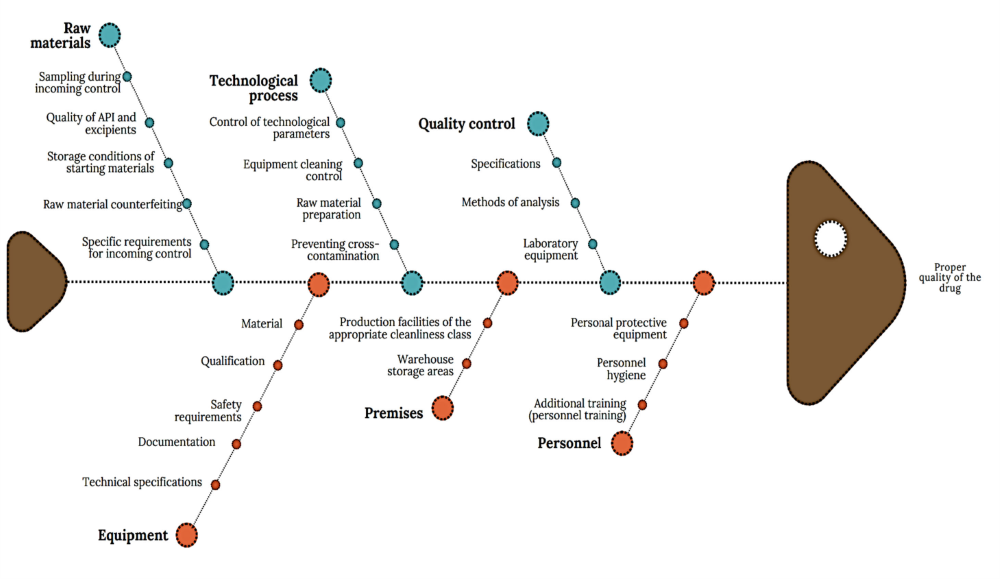
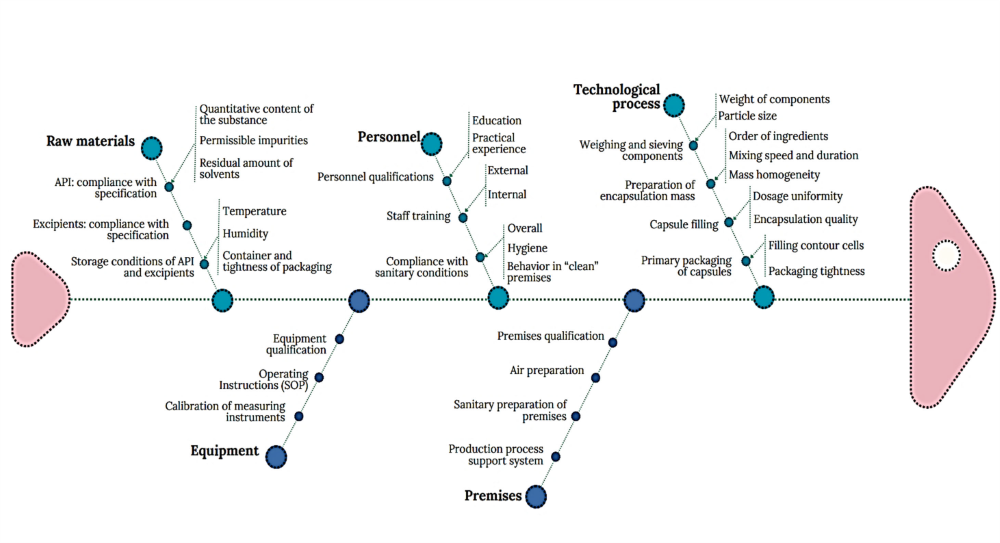
The general Ishikawa diagram, which allows taking into account the multilevel nature of factors affecting the quality of a drug as a final product and establishing key links between them, is shown in Figure 3. This diagram is based on the requirements of current legislation, WHO recommendations and national standards [11, 12].
According to the diagram (Fig. 3), the main risk factors of drug production include the quality of raw materials, technological process, quality control, premises, equipment, personnel, which are detailed by level 2 factors. Among the listed main factors, it is possible to distinguish factors of the first and second order in terms of significance, establish their links and group them.
1. Risk Prioritization and Justification of Focus
To move from a mere listing of factors to a justified conclusion about their criticality, a semi-quantitative risk assessment was performed. An expert panel (n=5) scored each identified cause from the general diagram (Fig. 3) on scales of Probability (P) and Severity (S) as described in the Methods. The calculated Risk Priority Number (RPN) served as a quantitative filter. The distribution of high-priority risks (RPN ≥6) across categories is presented in Fig. 3. As evidenced, the “Raw Materials” and “Technological Process” categories contained over 70% of all high-priority risks. This objective data directly supports our decision to focus the subsequent detailed analysis precisely on these areas, as presented in Tables 1 and 2 and in the adapted diagram (Fig. 4).
2. Detailed Analysis of High-Priority Risks
Following the prioritization, the most critical risks were analyzed in depth considering the peculiarities of production processes and the influence of factors on the quality of the finished drug, they can be divided into specific and general. Specific factors include the quality of the raw materials, quality control of the raw materials, intermediate and finished products, and the technological process. It is these factors that were subjected to the most thorough analysis during our study.
Of course, the main influence on the quality of the future drug is the quality of the raw material. Its quality risks are characterized by several secondary factors, illustrated in the diagram.
During the quality control of the finished drug, the choice of the API/excipient and the development of a method for its qualitative and quantitative determination, as well as the assessment of the microbiological state, are of great importance. All specification parameters are developed in accordance with the general guidance on finished drugs.
When conducting stability tests for drugs with several APIs, the possible mutual influence on the process of qualitative and quantitative determination of substances should be considered. Validation of the methods used is an urgent and mandatory task in the development of the MCQ.
Fig. 3. Diagram of cause-and-effect factors for ensuring the quality of medicines ↓
Factors such as premises and personnel have an unconditional impact on ensuring the quality of medicinal products. However, given the standardized requirements for both personnel and premises set out in the licensing conditions, we decided to specify only specific requirements for these factors. The main research was devoted to the input raw materials, technological process, quality control and equipment, which are decisive in the process of pharmaceutical development and introduction of new original medicinal products.
Production premises should follow a logical sequence of the production process. For both APIs and excipients, temperature and humidity control, filtration and effective ventilation are of great importance. Weighing of raw materials should be carried out in a separated from the production process premise. The design of the premises should facilitate compliance with special measures to prevent cross-contamination and facilitate cleaning when dust is generated (sampling, weighing, grinding, mixing, sieving and other operations). Warehouses should have sufficient capacity for orderly storage of different categories of raw materials. Containers should be placed in such a way as to ensure free air circulation. The storage area should provide protection of materials from weather conditions and control of proper storage conditions. Warehouses should have separate areas: for quarantined materials, approved materials and rejected starting materials.
Production personnel and quality control personnel must have special training. The authorized person must have specific knowledge of the processing and quality control of materials in the production of film-coated tablets. Strict adherence to hygiene requirements by personnel during the production process prevents further contamination of raw materials, intermediate products and finished products. Production personnel who are in direct contact with raw materials must be provided with personal protective equipment: masks, gloves, glasses. Persons with allergic diseases or predisposition to them are not allowed to work with raw materials.
Pharmaceutical equipment used must meet the requirements of good manufacturing practice. The operational characteristics of the equipment must ensure the performance of the production task within the operating load range. The technical characteristics of the equipment must correspond to the planned range and volumes of product output. The material of the equipment must not interact with the raw materials or products, serve as a source of contamination of the intermediate and finished product.
Qualification of critical equipment, process validation and change control ensure the reproducibility of the production process, thereby guaranteeing consistent quality and efficacy of different batches of the drug produced.
In order to minimize the potential risks of industrial production of hard capsules under the conditional name “Praziquantel Plus” when extrapolating laboratory technology, their analysis was conducted.
The main categories defined are raw materials, production process, equipment, personnel and premises. Particular attention was paid to potential risks associated with APIs and excipients (Table 1) and with the technological process (Table 2).
Taking into account the peculiarities of adapting laboratory technology to industrial conditions for the production of hard capsules under the conditional name “Praziquantel Plus”, the Ishikawa diagram focused on the peculiarities of the production process of this particular drug was constructed (Fig. 4). The categories “Raw materials”, “Technological process” and “Personnel” contain subcategories of the first and second order, maximally corresponding to the peculiarities of the production process of this drug.
The proposed Ishikawa diagram for hard capsules under the conditional name “Praziquantel Plus” can be used in the future in the following situations:
1. Analysis of the causes of product defects. If there is a problem with the quality of the capsules being produced (for example, capsules with uneven dosage), the diagram will help identify possible sources of the problem.
2. Quality control. The diagram will help to adjust the various stages of the production process to avoid defects at all stages of production.
3. Process optimization. Analyzing all possible causes of failures or costs during production allows to increase efficiency and reduce costs.
Fig. 4. Cause-and-effect diagram of quality assurance factors for hard capsules under the conditional name “Praziquantel Plus” ↓
The general cause-and-effect diagram constructed for our study identified six primary categories: Raw Materials, Technological Process, Quality Control, Premises, Equipment, and Personnel. This categorization is a direct operationalization of the integrated approach required by a PQS, as described in the ICH Q10 guideline. Furthermore, our systematic approach to risk identification, assessment, and control is fundamentally based on the principles of ICH Q9 (Quality Risk Management). While ICH Q9 provides the high-level framework, our study demonstrates its practical implementation by selecting an appropriate tool (Ishikawa diagram) for a specific, defined scope – the scale-up of a capsule product.
Our identification of raw material quality as a first-order risk factor (Table 1) is a universally acknowledged principle in Good Manufacturing Practice (GMP). The WHO in its GMP guidelines explicitly states that the quality of a finished product depends heavily on the quality of the starting materials.
Our detailed breakdown of the technological process resonates strongly with the findings of other researchers who have applied risk management to solid dosage forms. For instance, a review by Yu et al. (2014) [13] on the application of Quality by Design (QbD) to solid oral dosage forms identifies “blend uniformity” and “powder flow properties” as critical process parameters for capsule filling and tablet compression, which are directly related to our identified risks of “Heterogeneity of the encapsulation mass” and “Poor flowability of the mixture”. While Yu et al. approached this from a QbD perspective, our use of the Ishikawa diagram arrived at the same critical parameters, demonstrating the tool’s effectiveness in pinpointing areas that require rigorous control.
Similarly, the risk we identified concerning the “Weighing and Sieving” stage, particularly, inaccuracy in weighing, is highlighted in GMP literature as a common source of error. The PIC/S GMP Guide dedicates an entire annex to the Guidance on Good Manufacturing Practice for Medicinal Products, where it emphasizes that weighing, measuring, and other equipment used for critical steps should be calibrated and checked at defined intervals (5, Annex 4) [14].
Our analysis of the “Personnel” and “Premises” categories, while noting their standardized requirements, specified risks such as the need for personnel with special training and the exclusion of individuals with allergic predispositions. This aligns with the human factor engineering concepts increasingly integrated into GMP. The FDA's guidance on quality systems approaches emphasizes that as management should ensure that personnel have the education, training, and experience, or any combination thereof, to perform their assigned functions [15]. Our inclusion of a specific health screening (allergies) adds a nuanced, product-specific layer to this general personnel requirement, a detail that is often crucial in practice but may be absent from high-level guidelines.
Regarding “Premises”, our emphasis on a logical flow, separate weighing areas, and controlled warehouses to prevent contamination and mix-ups is a cornerstone of GMP facility design, as outlined by ISPE in its Baseline Guide: Oral Solid Dosage Forms [16]. Our use of the Ishikawa diagram effectively integrates these infrastructural and design requirements into the same risk assessment framework as the process parameters, promoting a holistic view of quality that is essential for successful pharmaceutical production.
Today, the pharmaceutical industry, following the automotive industry (AIAG-VDA FMEA Handbook 2019), often moves away from simple RPN multiplication in favor of Action Priority tables that take into account various combinations of S, O, D without arithmetic manipulations.
CONCLUSIONS
Contributors:
Semchenko K.V. – writing – original draft, supervision;
Iakovenko V.K. – methodology, writing – review and editing;
Shmalko O.O. – conceptualization, methodology – review and editing;
Oliinyk S.V. – conceptualization, writing – review and editing.
Funding. The research was conducted under the financing from the funds of National University of Pharmacy at the topic ща initiative direction “Development of composition, technology and biopharmaceutical research of medicines based on natural and synthetic raw materials” (state registration number: 0114U000945).
Conflict of interests. The authors declare that they have no conflict of interest in relation to this research, whether financial, personal, authorship or otherwise, that could affect the research and its results presented in this paper.
REFERENCES
Ключові слова: патентна монополія, «вічнозелені» патенти, лікарські засоби, зловживання патентними правами, конкуренція, інтелектуальна власність
Key words: patent monopoly, “evergreen” patents medicines, patent abuse, competition, intellectual property
Реферат
У статті здійснено систематизований аналіз наукових публікацій, присвячених сучасним тенденціям зловживання патентними правами у фармацевтичній галузі США, зокрема механізмам реалізації стратегій «вічнозелених» патентів та їх економіко-правовим наслідкам. Метою роботи було проведення систематизованого огляду наукової літератури щодо тенденцій зловживання патентними правами у фармацевтичному секторі США, з акцентом на механізмах реалізації стратегій «вічнозелених» патентів, їх економіко-правових наслідках та можливих напрямах удосконалення регуляторної політики. Для досягнення поставленої мети було застосовано методологію систематичного огляду літератури відповідно до рекомендацій PRISMA, у поєднанні із загальнонауковими та спеціальними методами наукового пізнання: системно-структурний, діалектичний, формально-логічний, порівняльно-правовий, методи аналізу та синтезу. Використання цих методів дозволило оцінити сучасний стан патентного регулювання в США, ідентифікувати правові інструменти продовження патентної монополії та проаналізувати їх вплив на доступність генеричних та/або біосимілярних препаратів. Матеріальною базою дослідження стали наукові публікації, аналітичні та статистичні звіти (PubMed, JAMA, Scopus, Springer, BMC, Oxford Academic), звіти та прогнози міжнародних організацій (DrugPatentWatch, ProClinical, PhRMA, Statista), а також офіційні документи Управління з патентів і торговельних марок США (USPTO), Угоди TRIPS та законодавство США у сфері патентного права (U.S. Patent Act, Hatch–Waxman Act). Із 286 ідентифікованих публікацій до якісного аналізу було включено 40 джерел, за період 2008–2025 років. Критеріями включення були джерела, що містять дані про патентний захист лікарських засобів, стратегії «вічнозелених» патентів, економічні наслідки та доступність медичних технологій. Джерела без прямого відношення до теми дослідження або з недостатньою науковою достовірністю виключалися. За результатами дослідження засвідчено, що фармацевтичні компанії США системно застосовують багаторівневі патентні стратегії, спрямовані на пролонгацію ринкової ексклюзивності після завершення базового патентного строку, підвищення цін на лікарські засоби та обмеження доступності терапії для споживачів. Водночас емпіричними даними підтверджено, що завершення строку дії патентів створює умови для активізації ринкової конкуренції та зниження цін на генерики на 30-50% протягом перших років. Підкреслено необхідність удосконалення регуляторних механізмів патентного законодавства США, включаючи обмеження практик «вічнозелених» патентів та забезпечення прозорості ринкових процесів. Надано прогноз щодо структурних змін у фармацевтичному ринку США в період 2025-2029 років, пов’язаних із завершенням строків дії ключових патентів на оригінальні лікарські засоби, а також запропоновано шляхи досягнення балансу між інтересами інноваційного розвитку та суспільними потребами в доступних лікарських засобах.
Abstract
Trends in the abuse of patent rights for medicinal products in the United States (review). Kodynets A.O., Doroshenko O.F., Volynets I.P., Dorozhko G.K., Petrenko V.O., Fedorova N.V. The article provides a systematic analysis of scholarly publications addressing contemporary trends in the abuse of patent rights in the pharmaceutical sector of the United States, with particular emphasis on the mechanisms for implementing “evergreen” patent strategies and their economic and legal consequences. The purpose of the study was to conduct a systematic review of the scientific literature on trends in the abuse of patent rights in the U.S. pharmaceutical sector, focusing on the mechanisms of “evergreen” patent strategies, their economic and legal implications, and possible directions for improving regulatory policy. To achieve this objective, a systematic literature review methodology was applied in accordance with the PRISMA guidelines, in combination with general scientific and special research methods, including the systemic-structural, dialectical, formal-logical, and comparative legal approaches, as well as methods of analysis and synthesis. The use of these methods made it possible to assess the current state of patent regulation in the United States, identify legal instruments for extending patent monopolies, and analyze their impact on the availability of generic and/or biosimilar medicines. The empirical basis of the study consisted of scientific publications, analytical and statistical reports indexed in PubMed, JAMA, Scopus, Springer, BMC, and Oxford Academic, reports and forecasts of international organizations (DrugPatentWatch, ProClinical, PhRMA, Statista), as well as official documents of the United States Patent and Trademark Office (USPTO), the TRIPS Agreement, and U.S. patent legislation (the U.S. Patent Act and the Hatch–Waxman Act). Out of 286 identified publications, 40 sources published between 2008 and 2025 were included in the qualitative analysis. Inclusion criteria comprised sources containing data on pharmaceutical patent protection, “evergreen” patent strategies, economic consequences, and access to medical technologies. Sources lacking a direct connection to the subject of the study or failing to meet scientific reliability standards were excluded. The results of the study demonstrate that U.S. pharmaceutical companies systematically employ multi-layered patent strategies aimed at prolonging market exclusivity beyond the expiration of the basic patent term, increasing drug prices, and restricting access to therapy for consumers. At the same time, empirical evidence confirms that patent expiration creates conditions for intensified market competition and a reduction in generic drug prices by 30–50% within the first years following market entry. The study emphasizes the need to improve regulatory mechanisms within U.S. patent law, including limiting “evergreen” practices and ensuring greater transparency of market processes. A forecast of structural changes in the U.S. pharmaceutical market for the period 2025-2029 is provided, associated with the expiration of key patents for original medicines, and approaches are proposed to achieve a balance between the interests of innovative development and societal needs for affordable medicines.
Інтелектуальна власність у фармацевтичній галузі виступає ключовим інструментом стимулювання інноваційної активності, забезпечення відтворюваності науково-дослідного циклу та збереження конкурентоспроможності на ринку. Як зазначають R.A. Pires, J.J. Ferreira, патентні механізми у сфері фармацевтики відіграють системоутворювальну роль в поєднанні стратегій інноваційного розвитку та інтелектуальної власності, створюючи своєрідний баланс між суспільними інтересами та приватними інвестиціями [26]. Патентний захист лікарських засобів (далі також – препарати/ ліки) традиційно обґрунтовується необхідністю компенсації значних фінансових витрат і тривалих клінічних досліджень, які супроводжують створення нових препаратів.
За оцінками аналітичних звітів DrugPatentWatch, середня вартість розроблення інноваційного лікарського засобу – від початкових лабораторних досліджень до впровадження продукту на ринок – становить близько 2,3 млрд дол. США, тоді як залежно від фармакологічної групи, технологічної складності та терапевтичного спрямування цей показник може коливатися в діапазоні від 300 млн до 4,5 млрд дол. США [39].
Водночас протягом останніх десятиліть спостерігається стійка тенденція використання патентних механізмів не лише як засобу стимулювання інновацій, але і як інструменту подовження монопольного контролю над фармацевтичним ринком. У науковій літературі таку стратегію позначають терміном «evergreening», або «вічнозелені» патенти, що передбачає створення багатошарових патентних портфелів, спрямованих на ускладнення або відтермінування доступу генеричних та/або біосимілярних лікарських засобів [1, 15].
Дослідження свідчать, що тенденція у фармацевтичній галузі полягає в зростанні кількості патентних заявок на незначні модифікації вже наявних препаратів – зміну рецептури, солі, ізомерів, поліморфів чи комбінування відомих лікарських сполук. При цьому кількість дійсно нових хімічних субстанцій, що впроваджуються у фармацевтичну практику, залишається стабільно низькою, демонструючи тенденцію до скорочення [3]. Таке зміщення акцентів з розроблення принципово нових активних речовин на мінімально модифіковані варіанти вже наявних молекул свідчить про інституціоналізацію стратегій «вічнозелених» патентів як поширеного бізнес-інструменту підтримання ринкової ексклюзивності.
Вказані стратегії зумовлюють низку економіко-правових наслідків, зокрема підвищення вартості лікування, зниження доступності життєво необхідних препаратів, уповільнення розвитку ринку генериків та зростання фінансового навантаження на системи охорони здоров’я. Означені процеси набувають особливої актуальності в умовах глобального зростання попиту на інноваційні лікарські засоби.
Мета дослідження полягає у проведенні систематизованого огляду наукової літератури щодо тенденцій зловживання патентними правами у фармацевтичному секторі США, з акцентом на механізмах реалізації стратегій «вічнозелених» патентів, їх економіко-правових наслідках та можливих напрямах удосконалення регуляторної політики.
МАТЕРІАЛИ ТА МЕТОДИ ДОСЛІДЖЕНЬ
Основою методологічного інструментарію дослідження стали загальнонаукові та спеціальні методи наукового пізнання: системно-структурний, діалектичний, формально-логічний, порівняльно-правовий, а також методи аналізу та синтезу. Вони забезпечили комплексний підхід до вивчення сучасних тенденцій зловживання патентними правами у фармацевтичному секторі США та визначення можливих напрямів удосконалення регуляторної політики.
Зокрема, порівняльно-правовий метод застосовувався для аналізу законодавчого регулювання патентного захисту лікарських засобів у США, а також для виявлення механізмів реалізації стратегій «вічнозелених» патентів і їх впливу на доступність лікарських засобів. Системно-структурний метод використовувався при оцінюванні структури фармацевтичного ринку, динаміки продажів оригінальних та генеричних препаратів, а також побудові прогнозів зміни цінової політики після завершення строку дії патентів. Методи аналізу та синтезу дозволили узагальнити наявні емпіричні та теоретичні дані щодо економічних та соціальних наслідків застосування стратегій «вічнозелених» патентів.
В основу дослідження увійшли матеріали з відкритих джерел, наукові публікації, статистичні та аналітичні звіти, нормативно-правові акти та міжнародні документи. Зокрема було проаналізовано матеріали з баз даних і пошукових систем (PubMed, JAMA, Scopus, Springer, BMC, Oxford Academic), звіти та прогнози міжнародних організацій (DrugPatentWatch, ProClinical, PhRMA, Statista), а також офіційні документи Управління з патентів і торговельних марок США (USPTO), Угоди TRIPS та законодавство США у сфері патентного права (U.S. Patent Act, Hatch–Waxman Act).
Теоретичною базою дослідження стали праці науковців та практиків різних галузей науки, зокрема у сфері фармацевтики та інтелектуальної власності, серед яких A. Kodynets, O. Doroshenko, I. Volynets, G. Dorozhko, V. Petrenko, V. Belitsky [1], S. Adepu, S. Ramakrishna [5], R. Aryan [7], M. Boldrin, D. Levine [8], H. Burke [9], G. Dwivedi, S. Hallihosur, L. Rangan [11], F. Gaessler, S. Wagner [12], L. Gorman [13], O. Granstrand, F. Tietze [14, 15]; C. Hemphill, B. Sampat [17, 18], M. Janczura, S. Sip, J. Cielecka-Piontek [20], S. Kolia [22], S. Myles [23], R. Pires, J. Ferreira [26], G. Quinn [27], P. Upadhyaya [35], V. Uyen [36], A. Vahrenwald, G. Pedde [37], F. Waller [38] та інші. Зокрема доповнимо, що роботи M. Boldrin, D. Levine, C. Hemphill, B. Sampat аналізують економічні ефекти патентної монополії; дослідження A. Kodynets, O. Doroshenko, I. Volynets та ін. розкривають правові аспекти патентного регулювання у фармацевтичній сфері; праці G. Quinn, O. Granstrand, F. Tietze присвячені інноваційним стратегіям фармацевтичних корпорацій та механізмам захисту інтелектуальної власності. Проте, незважаючи на наявність значної кількості досліджень, окремі аспекти тенденції зловживання патентними правами на лікарські засоби в США потребують подальшого вивчення.
Дослідження проведено у формі систематичного огляду літератури відповідно до рекомендацій PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses). Пошук джерел здійснювався у вищезазначених базах даних PubMed, Scopus, Web of Science, SpringerLink, Oxford Academic та інших відкритих джерелах за період 2008-2025 років з використанням ключових слів: pharmaceutical patents, evergreening strategies, patent abuse, generic drugs, Hatch-Waxman Act.
У результаті первинного пошуку ідентифіковано 286 джерел. Після видалення дублікатів (n=74) та скринінгу за назвами й анотаціями (n=212) було виключено 142 джерела, що не відповідали тематиці дослідження. Повнотекстовий аналіз проведено для 70 публікацій, з яких 30 було виключено через відсутність прямого зв’язку з патентним регулюванням або недостатню достовірність. У підсумку до якісного аналізу включено 40 джерел. Критеріями включення слугували наукові статті, аналітичні огляди, статистичні звіти, нормативно-правові акти та офіційні документи, що стосуються патентного захисту лікарських засобів, стратегій «вічнозелених» патентів та економічних наслідків їх застосування. Критеріями виключення стали джерела, що не містили прямого зв’язку з патентним регулюванням фармацевтичної галузі або не відповідали вимогам наукової верифікованості.
Особлива увага приділялась аналізу патентних стратегій найбільших фармацевтичних корпорацій, наслідків завершення строків дії патентів та моделей виходу на ринок генеричних та/або біосимілярних препаратів. Для систематизації даних було складено таблицю щодо строків дії патентів на оригінальні лікарські засоби, обсяги продажів та стратегії компаній щодо продовження ринкової ексклюзивності.
РЕЗУЛЬТАТИ ТА ЇХ ОБГОВОРЕННЯ
Фармацевтичні патенти становлять наріжний камінь сучасної інноваційної екосистеми, оскільки саме вони створюють ключові стимули для фармацевтичних компаній інвестувати у складний, тривалий та фінансово витратний процес розробки нових лікарських засобів. Як зазначає дослідження PhRMA, середній цикл створення й виведення на ринок одного фармацевтичного продукту триває понад десять років і вимагає близько 2,6 мільярда доларів інвестицій від моменту відкриття активної речовини до отримання схвалення регуляторних органів [28].
В умовах відсутності патентного захисту компанії не мали б можливості компенсувати витрати на розробку, оскільки конкуренти могли б швидко відтворити новий препарат і реалізовувати його у вигляді генерика за значно нижчою ціною. Патенти, надаючи тимчасову монополію (зазвичай протягом 20 років з дати подання заявки), забезпечують власникові виключне право на комерційне використання винаходу. Вбачаємо, що правова ексклюзивність дає змогу фармацевтичним компаніям встановлювати ціни, які враховують не лише фактичні витрати на дослідження й розробки, а й високі ризики невдач, адже більшість потенційних лікарських засобів не проходять клінічні випробування. Як наголошують F. Gaessler, S.Wagner, отриманий у результаті комерціалізації прибуток виконує подвійну функцію: по-перше, фінансує майбутні інновації, а по-друге, компенсує значний рівень відтоку ресурсів у процесі фармацевтичних досліджень [12].
Водночас патентна система у фармацевтичній галузі не позбавлена критики. Науковці звертають увагу, що патенти можуть призводити до надмірного зростання цін на лікарські засоби, обмежуючи доступ до необхідних медикаментів, особливо в країнах з низьким і середнім рівнем доходу. Як підкреслюють A. Vahrenwald, G. Pedde, випадки високої вартості ліків для лікування рідкісних (орфанних) хвороб демонструють напруження між необхідністю забезпечення стимулів для інновацій та суспільним обов’язком гарантувати доступність медичних технологій для всіх верств населення [37].
З аналогічних позицій виступають M. Boldrin, D. Levine, які у своїй фундаментальній праці «Against Intellectual Monopoly» стверджують, що надмірна монополізація інтелектуальної власності може, навпаки, гальмувати інноваційні процеси. На думку авторів, надлишковий патентний захист у фармацевтичній сфері формує бар’єри для конкуренції та обмежує потенціал подальших наукових відкриттів, зменшуючи загальну ефективність інноваційної системи [8].
Таким чином, хоча патентний механізм залишається основним інструментом підтримки інновацій у фармацевтичному секторі, його ефективність залежить від збалансованості між приватними економічними інтересами та суспільними цілями, зокрема забезпеченням доступності лікарських засобів і зниженням вартості лікування для споживачів.
Система патентного права США традиційно вважається однією з найбільш сприятливих для власників фармацевтичних патентів. Вона функціонує на основі положень Закону США «Про патенти» (U.S. Patent Act) [33] та тлумачиться в контексті положень Угоди про торговельні аспекти прав інтелектуальної власності (TRIPS, 1994) [6], ухваленої в межах Світової організації торгівлі (WTO), а також адміністративної практики Відомства з патентів і торговельних марок США (USPTO) [24]. Така правова архітектура створює передумови для формування багаторівневої системи охорони одного й того самого лікарського засобу, що забезпечує тривале збереження ринкової ексклюзивності інноваційних препаратів [7, 40].
Ухвалення в 1984 році Закону Хетча–Ваксмана (Hatch-Waxman Act) було спрямоване на стимулювання виходу на ринок генеричних лікарських засобів, проте водночас надало фармацевтичним компаніям-виробникам оригінальних препаратів низку правових механізмів для пролонгації строку дії патентної охорони [16, 18]. До таких інструментів належать подання нових заявок на патенти, що стосуються модифікованих версій уже наявних препаратів (зміни у складі, дозуванні, способі доставки активної речовини тощо), можливість ініціювання 30-місячних відстрочок розгляду заявок на генерики в межах судових процесів, що подаються відповідно до пункту IV Закону Хетча-Ваксмана [7].
Подібна правова модель піддається критиці з боку науковців та експертів фармацевтичного ринку, оскільки створює умови для формування так званих «патентних чагарників» (patent thickets) – складних мереж взаємопов’язаних патентів, які суттєво ускладнюють вихід генеричних аналогів на ринок [30, 34, 7].
Великі транснаціональні фармацевтичні корпорації формують розгалужені патентні портфелі, що охоплюють тисячі об’єктів інтелектуальної власності та забезпечують комплексний правовий захист розроблених ними лікарських засобів. Така стратегія дозволяє не лише зберігати домінуючі позиції на світовому ринку, але й ефективно контролювати доступ конкурентів до виробництва генеричних аналогів. Як зазначають A. Vahrenwald, G. Pedde, ключові фармацевтичні компанії – Pfizer, Merck & Co., Johnson & Johnson та інші – інвестують мільярди доларів у дослідження та розробки, отримуючи при цьому значні прибутки від комерціалізації своїх інноваційних препаратів [37].
На переконання A. Vahrenwald, G. Pedde, подібна багаторівнева патентна стратегія не лише зміцнює ринкові позиції фармацевтичних гігантів, а й утруднює вихід генеричних компаній на ринок після завершення первинного строку дії патенту. У результаті формується своєрідна «патентна фортеця», яка фактично продовжує комерційну монополію оригінатора і впливає на динаміку цін та доступність лікарських засобів для споживачів [37].
Закінчення строку дії патентного захисту є критичним етапом життєвого циклу будь-якого фармацевтичного продукту, оскільки саме воно знаменує перехід від монопольного ціноутворення до умов відкритої ринкової конкуренції. Цей процес, відомий як втрата виключних прав, зазвичай відкриває шлях для виходу на ринок генеричних та/або біосимілярних препаратів, що суттєво змінює цінову динаміку та структуру прибутків у фармацевтичній галузі.
Дослідження L. Gorman демонструє, що після завершення строку дії патенту середня ціна на рецептурні лікарські засоби знижується в межах 38-48%, що зумовлено активізацією конкуренції з боку виробників генериків [13]. Така тенденція вказує на тісний зв’язок між режимом патентного захисту та рівнем доступності лікарських засобів для споживачів.
Більш масштабне міжнародне дослідження, проведене M. Serra-Burriel, N. Martin-Bassols, G. Perényi, охоплювало 505 препаратів, термін дії патентів на які закінчувався у восьми країнах – Австралії, Канаді, Франції, Німеччині, Японії, Швейцарії, Великій Британії та США. Автори встановили, що зниження цін після завершення дії патенту було статистично значущим упродовж восьмирічного періоду, причому найінтенсивніше падіння спостерігалося саме в США: у перший рік після втрати патентного захисту ціни зменшувалися на 32% (95% ДІ, 24-39%), а протягом восьми років – на 82% (95% ДІ, 71-89%) [10].
Крім того, результати економічного моделювання, проведеного цими авторами, показали, що неврахування виходу генериків на ринок може призводити до упереджених показників економічної ефективності лікарських засобів – від +40% до -40%, залежно від заданого сценарію. Показово, що врахування етапу втрати виключних прав є необхідним елементом оцінки вартості медичних технологій і має бути системно інтегрованим у процес ухвалення рішень у сфері охорони здоров’я.
У найближчій перспективі світовий фармацевтичний ринок очікує на масове завершення строків дії патентного захисту низки найбільш прибуткових лікарських засобів. Протягом 2025-2029 років спливання терміну ключових патентів на такі продукти зумовить глибокі структурні зміни в конкурентному середовищі галузі [9]. Зі свого боку, матиме комплексний вплив на стратегії інноваційного розвитку фармацевтичних корпорацій, інвестиційну активність, а також цінову політику та динаміку ринкових консолідацій у форматі злиттів і поглинань.
Втрати патентного захисту змушують виробників оригінальних препаратів адаптувати бізнес-моделі, орієнтуючись на диверсифікацію портфелів, виведення модифікованих форм препаратів (нових солей, ізомерів, комбінацій активних речовин) або перехід до біотехнологічних рішень і персоналізованої медицини. Одночасно активізуються процеси зміцнення позицій на зростаючих ринках через стратегічні партнерства, угоди з виробниками генериків та «вічнозелених» патентів, спрямовані на пролонгацію періоду ринкової ексклюзивності.
У таблиці наведено вибірку найприбутковіших оригінальних лікарських засобів, строки дії патентів на які спливають у період 2025-2029 років [9, 22], із зазначенням компаній-виробників, основних терапевтичних показань, а також типових стратегій реагування на втрату патентного захисту.
Назва лікарського засобу Компанія Рік (роки) закінчення патенту Основне призначення Обсяг продажу на прикладі 2023 р. Стратегія компанії EYLEA Regeneron / Bayer 2025–2026 Вікова макулодистрофія, діабетична ретинопатія 5,9 млрд дол. США (США) Диверсифікація доходів, розробка нових молекул PROLIA/XGEVA (Denosumab) Amgen 2025–2026 Метастази в кістки, остеопороз +9% річного зростання у 2023 р. Фокус на науково-дослідних та дослідно-конструкторських роботах (НДДКР) в онкології та імунології PREVNAR 13 Pfizer 2026 Вакцина проти пневмококу Багатомільярдні продажі з 2010 р. Масштабні інвестиції в НДДКР та придбання Seagen Inc. XARELTO (Rivaroxaban) Bayer / Johnson & Johnson 2026 Антикоагулянт, профілактика тромбозів €4,5 млрд (2022), понад 80 млн рецептів у США Розвиток напрямів генної та клітинної терапії IBRANCE (Palbociclib) Pfizer 2027 Рак молочної залози (HR+/HER2–) Зниження продажів на 13% у 2023 р. Розширення онкопортфеля (Array BioPharma, Seagen) TRULICITY (Dulaglutide) Eli Lilly 2027 Цукровий діабет II типу 17-й за обсягами продажів у світі (2023 р.) Перехід на препарат нового покоління Mounjaro ELIQUIS (Apixaban) Bristol Myers Squibb / Pfizer 2027–2029 Профілактика тромбозів Один з лідерів у своєму класі Судовий захист патенту, дитяча ексклюзивність KEYTRUDA (Pembrolizumab) Merck & Co. 2028 Імунотерапія раку (меланома, легені, лімфома тощо) $33,7 млрд (прогноз на 2028 р.) Розширення портфеля онкопрепаратів, придбання Harpoon Therapeutics OPDIVO (Nivolumab) Bristol Myers Squibb 2028 Імунотерапія раку $9,0 млрд (2023 р.) Розробка шести нових препаратів для компенсації втрат OCREVUS (Ocrelizumab) Roche 2028–2029 Розсіяний склероз CHF 6,38 млрд (2023 р.) Інвестиції в дослідження раку молочної залози та кардіометаболічних хворобТаблиця. Строки дії патентів на оригінальні лікарські засоби, які спливають у 2025-2029рр. ↓
(торговельна марка)
(ДВТ, ТЕЛА)
Наведені дані демонструють, що перехід до постпатентної фази для більшості компаній супроводжується переглядом інноваційних пріоритетів і посиленням конкуренції з боку виробників генеричних та/або біосимілярних препаратів.
Відтак патентна система у фармацевтичній сфері виконує подвійну функцію: з одного боку, вона стимулює НДДКР, формуючи економічні стимули для розроблення інноваційних лікарських засобів; з іншого – забезпечує механізм економічної віддачі від інновацій, що підтримує сталий розвиток галузі [5].
Разом з тим, на тлі зростання вартості досліджень і посилення конкуренції, спостерігається поширення практик «вічнозеленого» патентування – стратегії, спрямованої на продовження комерційної виключності лікарських засобів понад початковий строк дії базового патенту. Сутність цієї стратегії полягає в системному отриманні додаткових патентів на незначні модифікації, поступові вдосконалення або нові способи застосування вже відомих препаратів [29, 31].
Хоча деякі з таких удосконалень можуть мати клінічну значущість (наприклад, покращення біодоступності чи зручності застосування), у більшості випадків вони використовуються як інструмент пролонгації ринкової монополії без суттєвого наукового прориву [5, 29, 31].
Показовим прикладом є препарат Phesgo (підшкірна комбінація трастузумабу та пертузумабу), схвалений у 2020 році. Завдяки зручнішому способу введення він швидко витіснив внутрішньовенні форми, для яких уже існували біосиміляри, фактично нівелюючи очікуваний ефект конкуренції. Інший приклад – Lytenava (внутрішньоочна форма бевацизумабу), який отримав статус нового оригінатора та власний патентний і регуляторний захист, попри завершення охорони базової форми Avastin [32].
У науковій літературі [22] та патентно-аналітичних звітах виокремлюють кілька найпоширеніших форм прояву стратегій «вічнозелених» патентів: нові форми та способи дозування, нові методи застосування або показання, патентування поліморфів й ізомерів, комбіновані лікарські форми [5, 22, 29, 20, 31]. Узагальнений перелік похідних речовин, з яких отримують «вічнозелений» патент, включає: фармацевтичні форми відомої хімічної сполуки; комбінації декількох активних речовин, які є відомими хімічними сполуками; солі, складні та прості ефіри відомих хімічних сполук; поліморфи; оптичні ізомери (енантіомери) відомих хімічних сполук; сполуки за формулою Маркуша; селекційні винаходи; проліки; метаболіти [2, 4].
Питання «вічнозелених» патентів є предметом гострих наукових і суспільних дискусій. Прихильники цієї стратегії розглядають її як один із механізмів підтримання інноваційної динаміки фармацевтичної галузі, стверджуючи, що інкрементальні вдосконалення лікарських засобів сприяють підвищенню їхньої якості, безпечності та зручності застосування для пацієнтів. Водночас критики наголошують, що стратегії «вічнозелених» патентів уповільнюють конкуренцію, призводять до підвищення цін на лікарські засоби та обмежують доступ до більш доступних генеричних препаратів [14, 36]. Попри широку критику, «вічнозелене» патентування залишається типовою стратегічною відповіддю інноваційних компаній на економічні виклики, пов’язані з високими витратами на дослідження та коротким періодом ефективної патентної охорони. Як зазначає O. Granstrand [14], фінансовий тиск, спричинений необхідністю максимізувати прибутки протягом обмеженого «вікна ексклюзивності», формує замкнене коло: високі витрати на НДДКР → впровадження «вічнозелених» патентів → зростання кількості патентних спорів і формування «патентних чагарників» → затримка виходу генериків → підвищення кінцевих цін для споживачів.
Вищезазначену тенденцію підтверджують сучасні аналітичні дані. Дослідження організації I-MAK демонструє системність патентних практик, спрямованих на тривале збереження ринкової ексклюзивності. Так, препарат Humira (AbbVie), призначений для лікування ревматоїдного артриту, має 311 патентних заявок, з яких 165 надано охорону, що забезпечило понад 43 роки монопольного контролю. Препарат Keytruda (Merck), розроблений для онкологічних показань, має 180 заявок, з яких 78 чинні, що гарантує близько 37 років ексклюзивності. Аналогічно, Enbrel (Amgen) перебуватиме під патентним захистом майже 50 років, що у 2,5 раза перевищує базовий 20-річний строк [19; 25].
Отже, дискусія в сучасному науковому та регуляторному середовищі вже не стосується питання, чи застосовуватимуть фармацевтичні компанії стратегії «вічнозелених» патентів, а радше – яким чином можна досягти балансу між комерційною необхідністю підтримання інновацій та суспільним інтересом у забезпеченні доступності лікарських засобів.
Як зазначають закордонні експерти, зокрема F.J. Waller, патенти є допоміжним інструментом захисту для нових підприємств, тобто патенти не дають можливість іншим фармацевтичним компаніям користуватися новими технологіями, які на момент виникнення є передовими та сучасними. Водночас компанія-винахідник може збагатитись за рахунок видачі ліцензій і отримання роялті [38].
Окремі проблемні питання щодо патентування лікарських засобів відзначила Н. Г. Медведева, яка виявила, що дотепер перед розробниками лікарського засобу постає складний вибір: чи патентувати нову хімічну сполуку або породжувати «вічнозелений» патент, зокрема через відсутність рекомендацій щодо опису родових структур або структур Маркуша [2, 23, 35].
Ще однією відомою структурою, яку використовують американські винахідники, є формула винаходу Джепсона (Jepson claim), за якою у вступній частині описується рівень техніки, а в заключній – елементи новизни, пропоновані заявником [21]. G. Quinn категорично проти зазначеної структури в заявках, через те, що, по-перше, законодавство США і так дозволяє охороняти винаходи, які є удосконаленнями роботи інших (35 U.S. Code § 101. Inventions patentable), тобто не є новаторськими, а по-друге, відповідна формула дозволяє патентовласникові використовувати преамбулу для викладу елементів або етапів заявленого винаходу, що є загальноприйнятими або відомими, тим самим користь від нового винаходу є мінімальна [27].
Вартує уваги, на наш погляд, наукова праця вчених G. Dwivedi, S. Hallihosur, L. Rangan щодо «оновлення» як імітації (обману), яке використовується в патентному праві. Зокрема, автори наголосили, що хоча «вічнозелений» патент не є офіційно-правовим поняттям, він позначає численні способи патентовласників фармацевтичної продукції за допомогою патентних законів розширити свої монопольні привілеї на строки, які зазвичай дозволяються та визначені відповідними нормативно-правовими актами, особливо щодо лікарських засобів з високим рівнем прибутку [11]. Мається на увазі те, що фармацевтичні компанії зловживають патентною системою на власну користь та видозмінюють вже наявні лікарські засоби, випускаючи їх в обіг як нові. У такий спосіб, отримуючи новий патент на оригінальний лікарський засіб, виробники навмисно обмежують випуск генеричних версій, запобігаючи виникнення конкуренції. Тому відповідна стратегія є недобросовісною.
Ефект, на який розраховують фармацевтичні компанії від власних дій, направлених на отримання «вічнозеленого» патенту, – це довготривале збереження ексклюзивності даних оригінального лікарського засобу, утримання монополії, а також максимізація прибутків від продажу [11]. Проте для споживачів масштабований випуск дороговартістних лікарських засобів, так званих «блокбастерів», не покращить здоров’я, а навпаки – чим дорожчий товар, тим менше можливості його придбати (особливо для третіх країн). Натомість очікуваний дохід від прибутків з часом може досягнути свого плато.
Ще одна робота C. Hemphill, B. Sampat свідчить про наявність іншої проблематики у створенні «вічнозелених» патентів. Зокрема, проведене дослідження підтверджує, що штучне продовження патентного захисту спричиняє виникнення патентів низької якості (lower quality patents) через збереження патентів на лікарські засоби, термін дії яких спливає. У перспективі, за висновками авторів, відбудеться стрімке усунення генеричних версій лікарських засобів, що може позначитись на здоров’ї населення по всьому світу [17].
Таким чином, результати систематичного огляду показали, що в США сформувалася інституціоналізована модель багаторівневого патентного захисту фармацевтичних інновацій, яка дозволяє компаніям пролонгувати ринкову ексклюзивність далеко за межі базового 20-річного строку. Аналіз відібраних джерел підтвердив, що стратегії «evergreening» є поширеною корпоративною практикою, яка реалізується через патентування нових форм, способів дозування, комбінацій активних речовин, ізомерів, поліморфів та нових показань. Емпіричні дані одностайно свідчать, що завершення строку дії патентів супроводжується істотним зниженням цін на лікарські засоби, що підтверджує безпосередній вплив патентного режиму на доступність лікування.
ВИСНОВКИ
Внески авторів:
Кодинець А.О. – концептуалізація, ведення, написання – рецензування та редагування, візуалізація, перевірка;
Дорошенко О.Ф. – ведення, редагування, формальний аналіз, перевірка;
Волинець І.П. – дослідження, методологія, курація даних, написання – початковий проєкт;
Дорожко Г.К. – формальний аналіз, ведення, перевірка;
Петренко В.О. – адміністрування проєкту, редагування, підготовка роботи до друку;
Федорова Н.В. – ведення, перевірка.
Фінансування. Дослідження не має зовнішніх джерел фінансування.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів.
REFERENCES
 UK
UK  EN
EN 